A Computational Approach to Identify Small Molecules Interact with the Crystal Structure of Programmed Cell Death Protein 1 as Potential Therapeutics for Cancer Immunotherapy
Abstract:
Bibliography/Citations:
1. Siegel, R.L., K.D. Miller, and A. Jemal, Cancer statistics, 2020. CA Cancer J Clin, 2020. 70(1): p. 7-30.
2. Turajlic, S., M. Gore, and J. Larkin, First report of overall survival for ipilimumab plus nivolumab from the phase III Checkmate 067 study in advanced melanoma. Ann Oncol, 2018. 29(3): p. 542-543.
3. Meng, X.Y., et al., Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des, 2011. 7(2): p. 146-57.
4. Crisan, L., S. Avram, and L. Pacureanu, Pharmacophore-based screening and drug repurposing exemplified on glycogen synthase kinase-3 inhibitors. Mol Divers, 2017. 21(2): p. 385-405.
5. Ngan, C.H., et al., FTSite: high accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics, 2012. 28(2): p. 286-7.
6. Jendele, L., et al., PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Res, 2019. 47(W1): p. W345-w349.
7. Krivák, R. and D. Hoksza, P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J Cheminform, 2018. 10(1): p. 39.
8. Shin, W.H., C.W. Christoffer, and D. Kihara, In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods, 2017. 131: p. 22-32.
9. Grosdidier, A., V. Zoete, and O. Michielin, SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res, 2011. 39(Web Server issue): p. W270-7.
10. Daina, A., O. Michielin, and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 2017. 7(1): p. 42717.
11. Dong, J., et al., ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J Cheminform, 2018. 10(1): p. 29.
12. Szklarczyk, D., et al., STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res, 2019. 47(D1): p. D607-d613.
Additional Project Information
Research Plan:
A. Rationale:
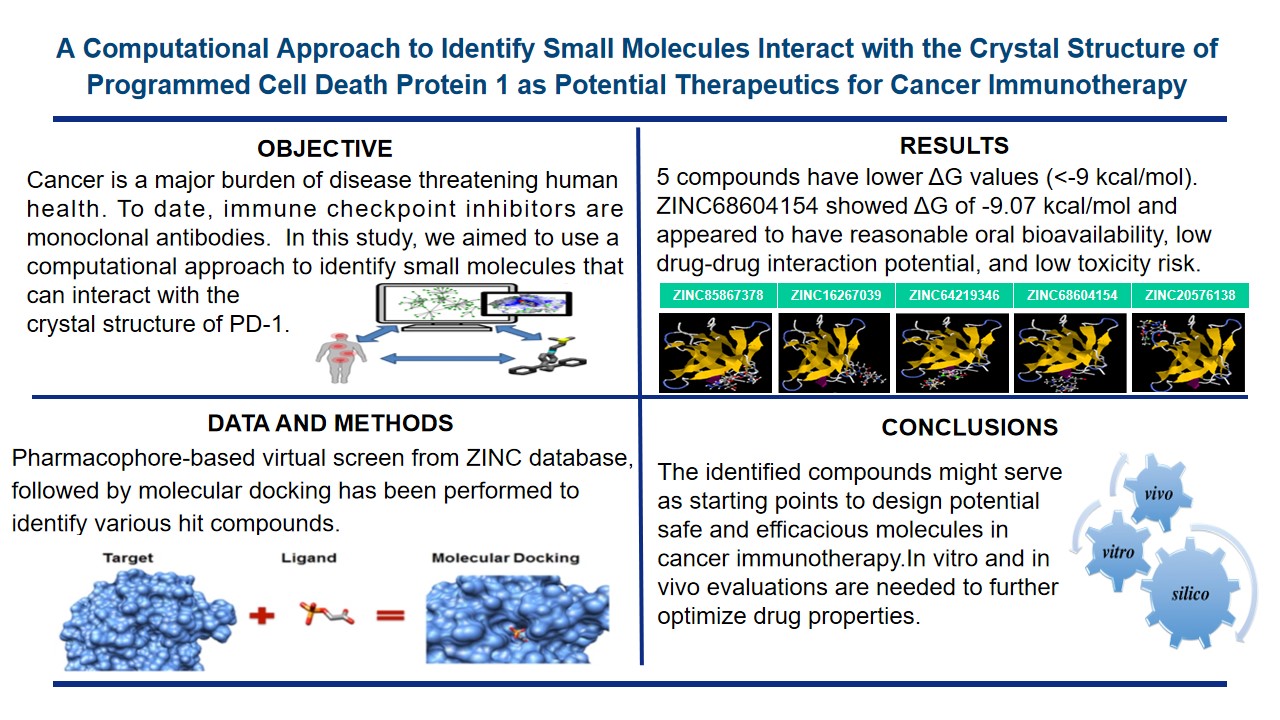
Globally, cancer is a major burden of disease threatening human health. In previous decades, the most common types of cancer treatments were surgery, radiotherapy, and chemotherapy. Recent advances in immunotherapy, a type of cancer treatment harnessing the immune system to fight cancer, established itself as one of the pillars of cancer treatment improving the prognosis of patients with different hematological and solid malignancies. Immune checkpoints (ICIs), which are a plethora of inhibitory mechanisms hardwired into the immune system, are important for modulating the duration and amplitude of physiological immune responses in tissues in order to limit collateral tissue damage. To date, immune checkpoint inhibitors (ICIs) are monoclonal antibodies that block immune checkpoints to augment T-cell–mediated tumor destruction. ICIs in current clinical settings target programmed death-1 (PD-1), programmed death-ligand 1 (PD-L1), or cytotoxic T lymphocyte antigen 4 (CTLA-4) are regarded as breakthroughs in cancer immunotherapy.
PD-1/PD-L1 pathway regulates the induction and maintenance of immune tolerance in the tumor microenvironment. Due to the inherent limitations of antibodies, it is reasonable to consider discovering orally bioavailable small molecule inhibitors that target programmed cell death protein 1/programmable death-ligand 1 (PD-1/PD-L1) signaling pathway as an alternative. Discovering a new therapeutic drug is a complex, costly and lengthy process. A combination of computational methods is an excellent replacement to identify potential drug candidates from the large compound libraries. There are huge unmet medical needs for the treatment of cancer. Leveraging biology to understand human diseases like cancer and using advanced technology has always fascinated me. I am very interested in gaining insight into cancer immunotherapy and to address real-world challenges and look for innovative ways to solve them while researching.
B. Research Questions:
1.) Are there binding sites for small molecule ligands on the PD-1 protein?
2.) Which small molecules likely bind to PD-1 protein with higher binding affinity and how to identify them?
3.) What the body does to these small molecules and what these small molecules does to body?
4.) What is the connection between the PD-1 protein and the CTLA-4 protein?
C. Aim:
The main objective of the project was to use a computational approach to identify the possible small molecules that can interact with PD-1, and further serve as starting points to design potential safe and efficacious compounds in cancer treatment.
Plan to achieve:
- Find potential binding sites on the PD-1 protein for small molecule ligands using various computer programs.
- Construct a pharmacophore map and utilize molecular docking to virtually screen thousands of small molecules to the ones that bind to PD-1 protein with higher binding affinity.
- Analyze the effectiveness of these small molecules in the body by looking at Lipinski’s Rule of Five and running absorption, distribution, metabolism, excretion, and toxicity (ADMET) tests.
- Analyze the interconnectivity between PD-1 and CTLA-4 proteins by also utilizing small molecule docking for CTLA-4 and constructing a protein interaction map, for the potential of targeting both proteins.
D. Methods and Procedures:
Materials and Data Collection
The research used crystal protein structures from the Research Collaboratory for Structural Bioinformatics (RCSB) database, I extracted the codes for both the structures of the PD-1 protein and of the PD-1/PD-L1 complex. The database gave me two codes 3RRQ for the PD-1 protein, and 3BIK for the complex.
Validation of Protein Structure
After I entered the PDB codes into the PROCHECK software in order to validate the crystal structures by looking at the torsion angles for the residues in the generated Ramachandran plots.
Searching for Various Binding Sites
I entered the PDB code for the PD-1 protein into three programs DogSiteScorer (a geometric method), FTsite (an energetic method), and PrankWeb (a machine learning based method) for the identification of possible ligand binding sites on the PD-1 protein.
PDB code was entered into the search box on the ProteinsPlus server and then press the “Go” button. Next, I chose DoGSiteScorer and pressed the “Calculate” button for running DoGSiteScorer with default settings for the results.
PDB code was entered into the search box on the FTsite server. Then inputted the job name and email address to receive a notification upon the completion of the job. Next, I pressed the “Find My Binding Site” button for running FTsite with default settings for the results.
PDB code has been entered into the search box on the PrankWeb server and then press the “Submit” button for running PrankWeb with default settings for the results.
Pharmacophore Query Construction using PocketQuery
PocketQuery is a great tool to use for the beginning of a pharmacophore screening. It allows for the identification of the most important protein residue clusters for ligand complexation. The chemical and structural features of these residue clusters are what make up the pharmacophore query.
I entered the PDB code into the search box on the PocketQuery server and then pressed the “Search” button for running PocketQuery with default settings for the results.
Pharmacophore Based Virtual Screening using ZincPharmer
The 6 highest ranked clusters obtained from PocketQuery were used as a query for pharmacophore-based virtual screening through ZincPharmer, which is an online platform (http://zincpharmer.csb.pitt.edu) for screening the commercially available compounds in the ZINC database. ZincPharmer used the query and produced a pharmacophore feature map which was then used in determining which small molecules from the ZINC database interacted best with the pharmacophore features. Narrowed thousands of ZINC compounds into list of 30.
Molecular Docking with PD-1
Selected hit compounds obtained by pharmacophore screening were subject to molecular docking, which was performed using SwissDock under the accurate mode (http://www.swissdock.ch). The program took in the protein code and the 30 small molecules to see how well they bound to each other. The results received were two energy values Full Fitness and Gibbs Free Energy. I decided to use the lowest Gibbs Free Energy because it is more generally understood and widely used.
Crystal structure of PD-1 (PID ID: 3RRQ) obtained from the protein data bank website was uploaded to SwissDock as “target selection”. Zinc ID identified by PocketQuery was entered as “ligand selection”. Next, I uploaded the converted mol2 file to SwissDock as “ligand selection”. I inputted the job name and email address to receive a notification upon the job completion. Then I pressed the “Start Docking” button to perform molecular docking for the results.
Molecular Docking with CTLA-4
More docking interactions of the selected molecules with CTLA-4 (PDB ID: 3OSK) were also explored using the same methods listed above.
Drug likeness property and ADME-tox prediction
Although these small molecules can bind well with the PD-1 protein I still needed to see how well they worked inside the body. To do this I looked at two aspects: Lipinski’s Rule of Five and ADMET tests.
I used the SWISSADME server to analyze the Drug-like properties of the selected molecules. After that, the ADME-tox evaluation for each of the molecules was conducted by using the online-based server, ADMETlab (https://admetmesh.scbdd.com/). For convenience interpolation, the numeric and categorical values of the results generated by the ADMETlab server were converted into qualitative values according to the documentation described online. The smiles format of each selected molecule was entered as input to SWISSADME or ADMETlab. Then I pressed the “submit” button to perform analysis with default settings for the results. The criteria included: water solubility, whether the small molecules satisfy Lipinski rules, bioavailability, and safety profiles (i.e., risk for liver toxicity).
Network analysis highlights non-random interconnectivity between PD-1 and CTLA-4
I also wanted to analyze the connection between the PD-1 protein and the CTLA-4 protein. To do this I constructed a protein interaction map seeing how many similar protein-protein interactions the two proteins had using the data in the STRING database.
E. Risk and Safety:
No statements of approval or informed consent will be required for this study as we obtained data from an open access database. No Identify any potential risks and safety precautions in this study.
Questions and Answers
1. What was the major objective of your project and what was your plan to achieve it?
The main objective of the project was to use a computational approach to identify the possible small molecules that can interact with PD-1, and further serve as starting points to design potential safe and efficacious compounds in cancer treatment.
Plan to achieve it:
1.) Are there binding sites for small molecule ligands on the PD-1 protein?
2.) Which small molecules likely bind to PD-1 protein with higher binding affinity and how to identify them?
3.) What the body does to these small molecules and what these small molecules does to body?
4.) What is the connection between the PD-1 protein and the CTLA-4 protein?
I developed a project research plan based on project goals, then executed the planned project steps to achieve goals.
a. Was that goal the result of any specific situation, experience, or problem you encountered?
Biology, medicine, and the health sciences have always been my favorite subjects throughout school. Dedicating my life to improving the lives of others is very appealing to me. As a result of the advancing research in recent years, interdisciplinary researches are more important for making major breakthroughs in health-related fields. For example, genomics is usually considered the study of DNA itself, whereas proteomics is broadly construed to represent the study of proteins expressed by genes. However, nowadays work in these fields requires more quantitative skills in mathematics and computer sciences, along with a thorough knowledge of the associated biology.
There are huge unmet medical needs for the treatment of cancer. Leveraging biology to understand human diseases like cancer and using advanced technology has always fascinated me. I am very interested in gaining insight into cancer immunotherapy and to address real-world challenges and look for innovative ways to solve them while researching.
b. Were you trying to solve a problem, answer a question, or test a hypothesis?
I was trying to solve a problem and answer a question. The question was whether there were possible small molecules that could bind to PD-1 and interfere with the protein pathway. The actual identification of those small molecules would be solving the problem.
2. What were the major tasks you had to perform in order to complete your project?
The major tasks I had to perform in order to complete the project included developing a research plan, literature search, data analysis and paper writing.
a. For teams, describe what each member worked on.
I independently conducted the research and wrote the entire paper with supervision from my mentor Dr.Moustafa Gabr. Dr.Gabr also guided me through how to utilize certain programs better. Also, I had another science mentor Dr.Wang who helped critique my paper at the end so that I would be able to make thorough improvements.
3. What is new or novel about your project?
a. Is there some aspect of your project's objective, or how you achieved it that you haven't done before?
Yes, the entire project was a new experience for me. Although I have had experience working on computer aided research projects, I have never done one looking for potential treatments and drugs. In addition, many of these programs I have never used before. For example, PocketQuery, ZincPharmer etc…
b. Is your project's objective, or the way you implemented it, different from anything you have seen?
Currently only monoclonal antibodies have been approved for immune checkpoint blockade therapy, so the objective of the project was to look for small molecules that could interfere with the PD-1/PD-L1 pathway. There have been studies looking for potential small molecules that interfere with this pathway in the past, most of them are still in the early development stage. In the current study, we use a computational approach to identify the possible new small molecules that can interact with PD-1, and further serve as starting points to design potential safe and efficacious compounds in cancer treatment.
c. If you believe your work to be unique in some way, what research have you done to confirm that it is?
I conducted a thorough literature search to confirm.
4. What was the most challenging part of completing your project?
The most challenging part about my project was related to understanding the computer programs. For example, trying to understand how to integrate the numerous software into my research plan, using multiple tools on the program and analyzing the different results different computer programs gave out.
a. What problems did you encounter, and how did you overcome them?
The PD-1/PD-L1 protein pathway was a new field to me and I was very lost at first when looking at the very complicated pathway. To tackle this, I needed to read a lot of background literature to understand the pathway, and to propose my research plan. However, sometimes the research plan was not supported by new background readings or didn’t capture the full picture of what I was looking for. As a result, I needed to rethink and modify my research plan based on new information I read and inputs from my mentor.
b. What did you learn from overcoming these problems?
This cycle of designing, testing, and failure is extremely important especially in science because it makes sure that the end product is supported by data and encapsulates the complete picture. The only way to make new scientific discoveries is by stepping into the unknown and to not be afraid of failure because I know that part of the journey is failure.
5. If you were going to do this project again, are there any things you would you do differently the next time?
If I were to do the project again, I would do two things differently. First, I would get a good grasp on the technical aspects behind the computer programs I use earlier, so that I would be able to make better sense of the data and plan my research procedure better. Second, I would read much more background information at the beginning so that later on I would understand the computer aided drug design better.
6. Did working on this project give you any ideas for other projects?
Yes, working on this project did give me ideas for other projects. For example, a next step, I would like to see if we could modify the small molecules to make them work better. For instance, make a small molecule follow all of Lipinski’s Rules or have better absorption, distribution, metabolism, excretion, and lower toxicity profiles.
7. How did COVID-19 affect the completion of your project?
I completed the research at home. I obtained data from an open-access database, conducted qualitative data analysis, and discussed with my mentor via zoom meetings. Covid-19 has had no major impact on my work.