DeepLPI: a novel deep learning-based model for protein–ligand interaction prediction for drug repurposing
Abstract:
Bibliography/Citations:
No additional citationsAdditional Project Information
Project files
Research Plan:
Rationale:
The time and financial cost of drug research and development have always been a big problem for pharmaceutical companies and patients. Especially in the case of the Covid-19 pandemic, developing new drugs may take too long to meet the patient’s needs. In order to lower the expenditure and development failure rate, repurposing existing and approved drugs by identifying interactions between drug molecules and target proteins based on computational methods have gained growing attention. DTI (Drug Target Interaction) is the most common method for computer-based drug repurposing. The method uses computers to predict the binding affinity between the drug molecules and protein sequence and further help to evaluate if the drug can be used for the virus. However, current DTI models cannot simultaneously attain high prediction accuracy and transferability. Some traditional machine learning models use human select features to make predictions. They have a huge amount of data to generalize the model, but their accuracy is insufficient. Some other models use highly precise 3D special data for prediction. The 3D special data helps the models to have very high accuracy, but because the data is only available in very limited situations, the training set is not large enough. So they don’t have a high transferability.
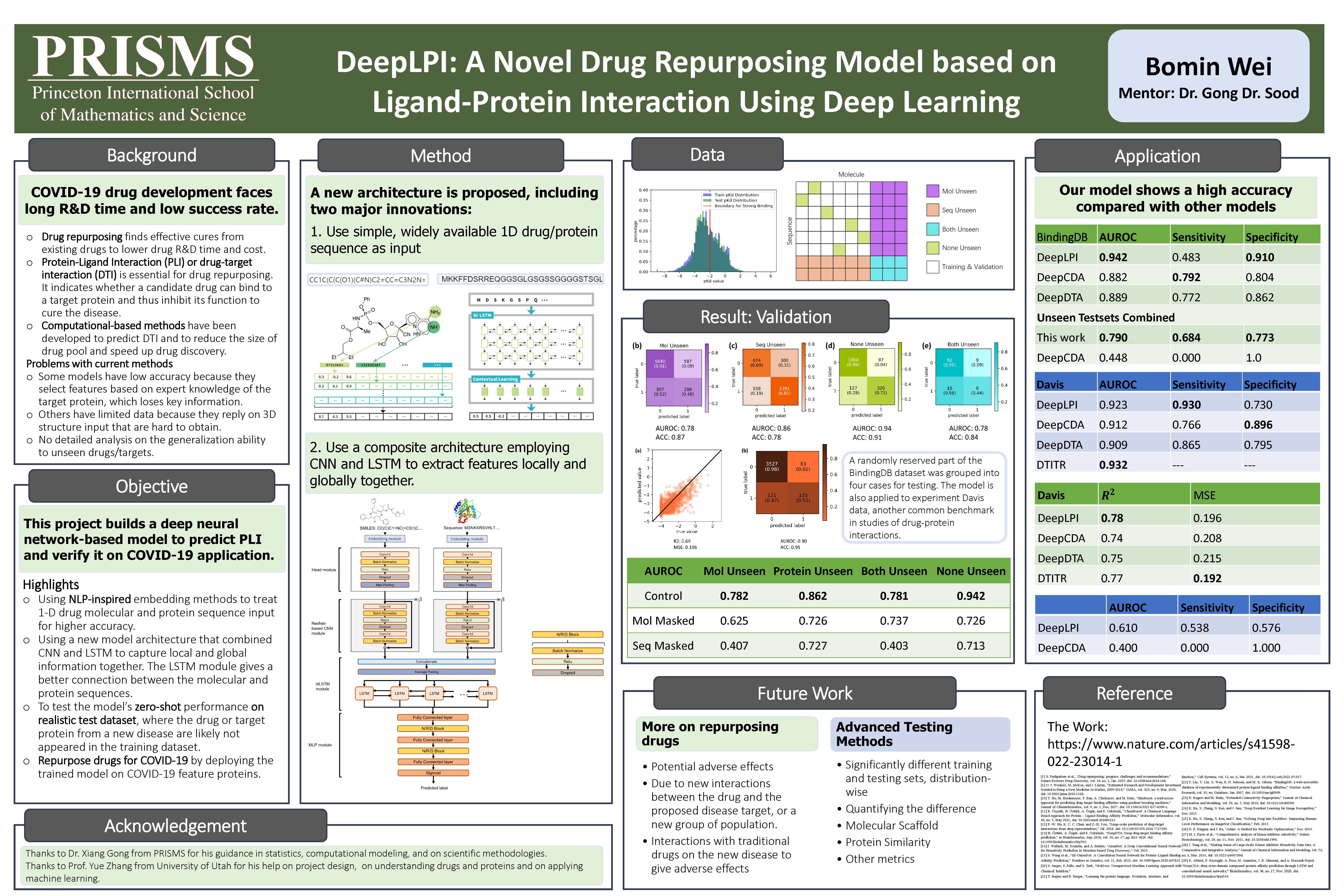
In this project, I am going to construct a new model called DeepLPI. The model will be able to achieves high accuracy and transferability at the same time. Instead of using 3-D structure data which is rarely available in databases, I propose to use 1-D protein (FASTA format, just like representing every amino acid in the chain as a single character) and molecular (SMILES, Simplified Molecular Input Line Entry System, format) data as the input for making the prediction. Inspired by AlphaFold 2 and some currently famous nature language processing models, the drug molecule and protein sequence data will be treated as language sentences: the language-like embedding (word2vec-based embedding architecture) and feature extraction (bi-LSTM and transformer) approach will be tested in the model.
Overfitting and virtual-high accuracy are the other enormous problem in DTI models. In this research, I will introduce several new methods for testing the model’s performance. First, the zero-shot testing dataset will be prepared for testing the real-life situation of the protein sequences that are never used during the training process. Second, the masked test that makes half the input the random value will be used to evaluate further the balance of weight on the drug molecule and protein sequence when evaluating the binding affinity. Third, several existing methods will be trained and tested using the same dataset for horizontal comparison to further evaluate my model’s performance.
Research Questions:
How can I build a model which can better extract features?
How to balance the difference in dimensions of embedding on drug molecule and protein sequence?
Are the modules we use actually helping with the result?
How to evaluate the model’s performance? (What specific quantification value can be used?)
How to split data to build the zero-shot test set? How can we use the existed dataset to simulate a real-life situation?
What can the result from the masked test show the model’s performance?
Procedures:
1. Setting up the computation node for model training and testing. Build up the Python environment and set up GitHub Repositories.
2. Research existing DTI articles and find an appropriate dataset for training and testing.
3. Research and implement embedding methods that convert drug molecule strings and protein sequences into numeric vector representations—testing different embedding methods if needed.
4. Setting up a basic model architecture by treading the drug and protein information separately to extract features first, then merging and making the prediction.
5. Decide the measurement values for testing the model’s performance. Then train and test the model on one example dataset. Revise the model network by considering adding the following modules into the model.
- Add LSTM layer for treading the final information matrix.
- Transformer may be used to first preprocess the embedded data.
- 1-D CNN module can be used to process the information matrix.
- Resnet Blocks might be useful.
- Pooling is the best way to reduce the size of vectors.
6. Optimize the model hyperparameter, including the size of different layers, the loss function, the learning rate, and the optimizer (method for gradient decent)
7. Doing further testing, including:
- Independent testing with a personal train-test splitting method. The method will split data into four sets, each representing a real-life situation.
- Transfer testing. Transfer the model into a Covid-19 real-life dataset to do the testing on if the model can correctly predict the performance of some drugs on Covid-19.
- Masked testing, which randomized half of the input. In this case, the model should not have the ability to correctly predict, or it could be overfitting.
- Comparing with existing methods with training and testing and the same dataset.
Safety:
This project involves only the use of computers and publicly accessible databases; therefore, the project does not have any risk or safety issues.
Questions and Answers
1. What was the major objective of your project and what was your plan to achieve it?
Due to safety and clinical studies requirements, new medicines often take 10-20 years to produce, which is inadequate for treating acute disorders like Covid-19. Repurposing approved medications is one solution, but testing their usefulness is challenging. Computer models are used to accelerate predictions, but they cannot attain high accuracy and transferability simultaneously. To break this situation, my research processed protein and molecule with a linguistic model and built a deep learning model to predict the binding affinity to accelerate the drug repurposing process.
a. Was that goal the result of any specific situation, experience, or problem you encountered?
The outbreak of Covid-19 made the schools close, and I had to attend classes online at home. I missed being in school with my friends and having in-person classes. So, I started thinking about what I could do to fight Covid-19. At that time, vaccination was a major hope for returning to normal life. To assist with vaccine development, I worked on a computational model that predicted the probability of virus mutation. The project was unsuccessful since the prediction could not change the result since vaccine development may take too long, and the prediction already lost efficiency. So, I changed my idea to focus on drugs that can cure patients more efficiently.
I was surprised to learn that developing a new drug may take about 10 to 20 years, which is too long to solve the emergent situation of Covid-19. I learned the interesting concept of drug repurposing that can accelerate the safety trial by reusing approved drug. But the strategy still have to test large amount of candidates, and the current computational and machine learning methods are not effective enough in virtual drug screening because they rely too much on the complex spatial structure.
Just at the same time, the world-shocking success of AlphaFold2 inspired me to start the new project that using Natural Language Processing (NLP) models for drug repurposing. Therefore, I took on the project and started to learn how to build deep neural networks and integrate NLP technique in prediction drug and protein interactions.
b. Were you trying to solve a problem, answer a question, or test a hypothesis?
The project is trying to solve the problem that current Drug develop process is slow and costly, and current DTI methods are not efficient enough for find usable existing drugs. I'm going to develop a deep learning model that can find the potential use of some existing drugs on new diseases.
2. What were the major tasks you had to perform in order to complete your project?
1. Setting up the computation node for model training and testing. Build up the Python environment and set up GitHub Repositories.
2. Research existing DTI articles and find an appropriate dataset for training and testing.
3. Research and implement embedding methods that convert drug molecule strings and protein sequences into numeric vector representations—testing different embedding methods if needed.
4. Setting up a basic model architecture by treading the drug and protein information separately to extract features first, then merging and making the prediction.
5. Decide the measurement values for testing the model's performance. Then train and test the model on one example dataset. Revise the model network by considering adding the following modules into the model.
1. Add LSTM layer for treading the final information matrix.
2. Transformer may be used to first preprocess the embedded data.
3. 1-D CNN module can be used to process the information matrix.
4. Resnet Blocks might be useful.
5. Pooling is the best way to reduce the size of vectors.
6. Optimize the model hyperparameter, including the size of different layers, the loss function, the learning rate, and the optimizer (method for gradient decent)
7. Doing further testing, including:
1. Independent testing with a personal train-test splitting method. The method will split data into four sets, each representing a real-life situation.
2. Transfer testing. Transfer the model into a Covid-19 real-life dataset to do the testing on if the model can correctly predict the performance of some drugs on Covid-19.
3. Masked testing, which randomized half of the input. In this case, the model should not have the ability to correctly predict, or it could be overfitting.
4. Comparing with existing methods with training and testing and the same dataset.
a. For teams, describe what each member worked on.
This was not a team project.
3. What is new or novel about your project?
This project has two novel contributions.
1. The project considered the ligand and molecular information as natural language sentences and processed them based on some NLP methods. The model's architecture is also designed using NLP modules, such as LSTM and transformer.
2. Another problem of current approaches is that they test their model only using random split. This project introduces a novel testing method for showing the model's performance in different conditions. (e.g., using existing drugs for new diseases or treating existing diseases using new drugs.) The Masking Testing Method is also introduced to test if the model is overfitting.
a. Is there some aspect of your project's objective, or how you achieved it that you haven't done before?
Before this project, I have zero experiments on building deep learning models. In order to do this project, I spend hundreds of hours testing many models, learning the principle, and trying every method I need in the research. Specifically, when I learned the function of Res-Net module, I built more than twenty different versions of model to test the module's performance when replacing the traditional CNN module.
b. Is your project's objective, or the way you implemented it, different from anything you have seen?
The project's objective is solving an emergent problem using computer models, which is common, but how I implement it is novel. In this project, I used the LSTM module to extract features locally and globally together and tried using transformers to rate and extract essential features. Both approaches were common in natural language processing models (such as ChatGPT, which is well known) but rarely used in cheminformatics. I tried this approach because when I looked at the FASTA amino acid chain file, I felt the entire RNA chain was just like a sentence. Each amino acid can be considered as a word, and connecting them can be the sentence. So why don't I just try to use the NLP modules to process them? Luckily, I tried and succeded.
c. If you believe your work to be unique in some way, what research have you done to confirm that it is?
The architecture of the model and testing method I designed in this project differs from others. Before starting the project, I read many papers (including papers of DeepDTI, DeepCDA, DTITR, SimBoost, AtomNet, and Se-OnionNet) about different approaches to DTI. They introduce many different interesting ideas for building the model. However, although many used SMILES to enter ligand molecules (such as DeepDTI and DTITR), none of the projects considered the input a natural language sentence. Besides, for the testing part, they are only using random split. The only special thing is that DeepDTI introduced its own testing measurement. In this case, I believe my testing method is also novel and unique.
4. What was the most challenging part of completing your project?
Since my work is introducing an approach for DTI, designing and optimizing a proper model architecture was the most challenging part. The deep neural network model has very complex architectures. A complex architecture may capture more features but, at the same lead to overfitting. Balance the complexity and possibility of overfitting is very important and difficult.
a. What problems did you encounter, and how did you overcome them?
1. Building a good model which can both have high prediction accuracy, which is competitive with models that use high dimensional inputs, and have high transferability that using only 1D molecular and protein sequence as input is one of the most challenging parts of the project. In order to build a model to accomplish this goal, I tried a lot of different methods when I was building the model architecture. I have built nine versions of the model in total: I tested to use many embedding methods, such as SeqVec, AllenNLP, and Prot2Vec; and compared the performance of different model architectures, such as adding a transformer in the head module and using LSTM on molecular and protein respectively after the CNN module. In fact, most of the approaches didn't work, but they helped me to take one step closer to the best model architecture. With testing all possible failures, …
2. Inside the model, the embedding part can give us a vector that indicates the character of molecular and protein. For instance, after embedding two similar proteins, the vectors we received may be near each other in the vector space to show that they are similar. It means that the model's accuracy is highly based on the embedding process. So, although we may have an excellent result when evaluating the model. The model may still not practice in real life because when making predictions on some unknown or new proteins, the embedding methods may not give us an accurate result. To solve this problem, I checked a lot of published embedding methods and tested them to improve my own model's universality.
3. Once when I discussed my project with a senior school friend majoring in biology, she told me that the way to predict without using the protein 3D structure is questionable - for a single protein sequence, there are a lot of different possible 3D structures. For each structure, the position and way that the molecular ligand binds on the protein are different, so this prediction is unrealistic in theory. However, I believe this can be solved. DeepMind published a protein structure prediction model called Alphafold 2, which allowed people to predict protein structure with sequence. The success of Alphafold 2 told me that we could predict with only the protein sequence. Even though the theoretical background of using 1D protein sequence to capture 3D spatial structure has not yet been solidly established, I found confidence in my proposal to find a way to extract information from the single protein sequence string that keeps most of the information we need for making the prediction.
4. One time, the model's performance hit a bottleneck that could not be improved. My research advisor suggested that I submit my work to conferences and introduce the project to professors or university students for advice. So I submitted the work to ISMB and IDWeek conference and fortunately got accepted for a poster presentation. I presented my work and received many valuable inspirations.
b. What did you learn from overcoming these problems?
Firstly, I learned in solving a research problem, taking the time and effort to do the test is more effective than hesitating for a better design. Plus, implementing and testing an idea is very rewarding and exciting.
Secondly, I realized in research work that a rigorous theory might solidly support many proposals. On the one hand, we should try our best to reason the design with existing theories to make sure it won't fail on obvious mistakes, and on the other hand, we should be courageous to take on the challenge and test out our proposal with careful implementations.
5. If you were going to do this project again, are there any things you would you do differently the next time?
If I have a chance to do the project again, I would like to investigate my evaluation process first. There is still room for improvement regarding prediction accuracy, especially when the model is applied to external datasets. This is because I spent too much time improving the model's performance in internal testing, or the random split testing, before I noticed that the testing method could be improved.
6. Did working on this project give you any ideas for other projects?
This project helped me to notice the potential of NLP models. So, in the future, I will focus on the principle behind those NLP methods, such as the principle of backpropagation for LSTM and the optimization of embedding.
7. How did COVID-19 affect the completion of your project?
The pandemic mildly influenced the working schedule at school and at my mentor professor's University, and therefore I had to adjust meeting schedules from time to time, but it wasn't a big problem. Mostly I communicated with the professor online, which was not impacted by the pandemic. The research is conducted on computers and remote cloud servers (commercial devices), and the pandemic didn't impact the data collection.