Navigating the Tumor Microenvironment: Identifying Novel Biomarkers in Non-Small Cell Lung Cancer Using Single-Cell Transcriptomics
Abstract:
Bibliography/Citations:
Bibliography:
Jovic, D., Liang, X., Zeng, H., Lin, L., Xu, F., & Luo, Y. (2022). Single‐cell RNA sequencing technologies and applications: A brief overview. Clinical and Translational Medicine, 12(3). https://doi.org/10.1002/ctm2.694
Kalainayakan, S. P., FitzGerald, K. E., Konduri, P. C., Vidal, C., & Zhang, L. (2018). Essential roles of mitochondrial and heme function in lung cancer bioenergetics and tumorigenesis. Cell & Bioscience, 8(1). https://doi.org/10.1186/s13578-018-0257-8
Melo, C. M., Vidotto, T., Chaves, L. P., Lautert-Dutra, W., Reis, R. B. D., & Squire, J. A. (2021). The role of somatic mutations on the immune response of the tumor microenvironment in prostate cancer. International Journal of Molecular Sciences, 22(17), 9550. https://doi.org/10.3390/ijms22179550
Mithoowani, H., & Febbraro, M. (2022). Non-Small-Cell lung cancer in 2022: A review for general practitioners in oncology. Current Oncology, 29(3), 1828-1839. https://doi.org/10.3390/curroncol29030150
Orr, J. C., & Hynds, R. E. (2021). Stem cell–derived respiratory epithelial cell cultures as human disease models. American Journal of Respiratory Cell and Molecular Biology, 64(6), 657-668. https://doi.org/10.1165/rcmb.2020-0440tr
Remark, R., Becker, C., Gomez, J. E., Damotte, D., Dieu-Nosjean, M.-C., Sautès-Fridman, C., Fridman, W.-H., Powell, C. A., Altorki, N. K., Merad, M., & Gnjatic, S. (2015). The non–small cell lung cancer immune contexture. A major determinant of tumor characteristics and patient outcome. American Journal of Respiratory and Critical Care Medicine, 191(4), 377-390. https://doi.org/10.1164/rccm.201409-1671pp
Rodak, O., Peris-Díaz, M. D., Olbromski, M., Podhorska-Okołów, M., & Dzięgiel, P. (2021). Current landscape of non-small cell lung cancer: Epidemiology, histological classification, targeted therapies, and immunotherapy. Cancers, 13(18), 4705. https://doi.org/10.3390/cancers13184705
Stuart, T., Butler, A., Hoffman, P., Hafemeister, C., Papalexi, E., Mauck, W. M., Hao, Y., Stoeckius, M., Smibert, P., & Satija, R. (2019). Comprehensive integration of single-cell data. Cell, 177(7), 1888-1902.e21. https://doi.org/10.1016/j.cell.2019.05.031
Tan, Z., Chen, X., Zuo, J., Fu, S., Wang, H., & Wang, J. (2023). Comprehensive analysis of scRNA-Seq and bulk rna-seq reveals dynamic changes in the tumor immune microenvironment of bladder cancer and establishes a prognostic model. Journal of Translational Medicine, 21(1). https://doi.org/10.1186/s12967-023-04056-z
Wang, C., Yu, Q., Song, T., Wang, Z., Song, L., Yang, Y., Shao, J., Li, J., Ni, Y., Chao, N., Zhang, L., & Li, W. (2022). The heterogeneous immune landscape between lung adenocarcinoma and squamous carcinoma revealed by single-cell RNA sequencing. Signal Transduction and Targeted Therapy, 7(1). https://doi.org/10.1038/s41392-022-01130-8
Additional Project Information
Research Plan:
Rationale:
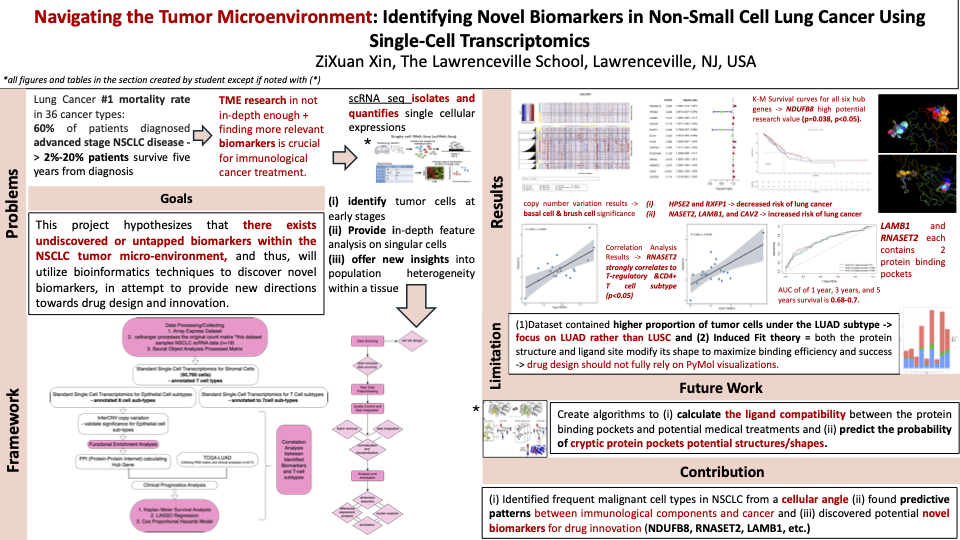
Non Small Cell Lung Cancer (NSCLC) represents eighty-five percent of Lung Cancer, the global leading cause of death, in which over sixty percent were diagnosed at a late-stage of the disease. The high degree of cellular heterogeneity in Non-Small Cell Lung Cancer (NSCLC) tumor formation calls for extensive exploration into tumor microenvironments (TME), indicating that TME research is underlooked in the status quo, consequently making the identification of novel genetic biomarkers a flagship for future cancer drug innovations. This study aimed to discover novel NSCLC by i) conducting standard procedure single cell RNA analysis towards both epithelial cell subtypes and immune cell subtypes, ii) utilizing clinical data and implementing statistical models to recognize potential genetic risk factor genes, iii) associating statistically significant NSCLC risk factor genes to potential cell subtypes, and iv) identifying novel biomarkers with protein binding pockets to allow for future drug design innovations. This project hypothesizes that there exists previously undiscovered or untapped biomarkers within the NSCLC tumor micro-environment, and thus, will utilize bioinformatics techniques to discover novel biomarkers, in an attempt to provide new directions towards drug design and innovation.
Research Questions:
- What is the optimal single cell transcriptomics method to analyze the dynamic between cancer cells and TME (tumor microenvironment)?
- How does frequently malignant cell types/subtypes contribute to identifying early stage NSCLC (Non-Small Cell Lung Cancer)?
- How does the immunological microenvironment indicate predictive patterns in cancer?
- Are there previously underlooked or untapped biomarkers, and how can bioinformatics techniques be employed to discover them?
- How can identifying potential novel genetic biomarkers aid the current bottleneck in drug discovery?
Hypotheses:
This project hypothesizes that there exists undiscovered or untapped biomarkers within the NSCLC tumor micro-environment, and thus, will utilize bioinformatics techniques to discover novel biomarkers, in an attempt to provide new directions towards drug design and innovation.
Procedures:
- Extract array express dataset from GEO database.
- Preprocess raw dataset with cellranger 7.1.0, namely, compare reference genomes, filter and correct data, and conduct process analysis on the human reference genome (GRch38).
- Utilize Seurat to perform quality control and data integration to filter out cells with less than 300 genes and over 10% of mitochondrial gene expressions.
- Employ functions in Rstudio to remove the batch effect, normalize, and standardize the dataset.
Data Analysis:
- Perform Cluster analysis on remaining cells with dimensionality reduction tools.
- Visualize Cluster analysis with the UMAP and Louvain algorithm.
- Annotate UMAP with marker genes found through various references.
- Repeat 5,6,7, for chosen immune cells.
- Repeat 5,6,7, for chosen epithelial cell sub-types.
- Use Infercnv to detect copy variation differences between epithelial cell sub-types and T-cells.
- Perform GO and KEGG functional enrichment analysis on differential expression genes.
- Calculate entities with low GSEA p-values.
- Visualize results for all three types of enrichment analysis.
- Use STRING to visualize the PPI.
- Utilize CytoHUbba to filter six hub genes, replying on the MCC algorithm
- Graph K-M Survival Curve based on matrix datasets and clinical sample information.
- Graph the change of expression levels of hub genes through time under two survival states and find statistically significant hub genes.
- Adopt the open-sourced TCGA dataset and conduct regression analysis to identify statistically significant risk genes. Further, the risk scores generated help to identify whether the correlation between the gene and cancer survival is positive or negative.
- Conduct LASSO regression analysis to confirm statistically risk variables.
- Graph the ROC curve to predict the 3 to 5 year survival rate of NSCLC patients based on the AUC values.
- Conduct Pseudotime Analysis with monocles and analyze the trends of the states of the T-cell subtypes and their relationship with NSCLC risks.
- Predict Protein Binding Pockets with Pymol and select genetic biomarkers useful for drug discovery.
Risk and Safety:
These procedures involve no wet lab design.
Questions and Answers
1. What was the major objective of your project and what was your plan to achieve it?
The main objective of my project was to identify novel genetic biomarkers, flagships for future cancer drug innovations. Specifically, I plan to discover novel new biomarkers by conducting standard procedure single cell RNA analysis towards both epithelial cell subtypes and immune cell subtypes, utilizing clinical data and implementing statistical models to recognize potential genetic risk factor genes, associating statistically significant NSCLC risk factor genes to potential cell subtypes, and identifying which biomarkers have protein binding pockets to allow for future drug design innovations.
a. Was that goal the result of any specific situation, experience, or problem you encountered?
I lost my grandfather to cancer. Although he had always followed an active and incredibly healthy lifestyle, he was diagnosed with stage 4 cancer when I was five– and the thought that someone as strong and robust as my grandfather could be defeated planted a seed for my obsession with curing cancer.
b. Were you trying to solve a problem, answer a question, or test a hypothesis?
I decided to focus my research on NSCLC because it accounts for 80%-85% of all deaths resulting from lung cancer, which holds the highest mortality rate within all 36 cancer types. Moreover, previous literature lacked emphasis on the tumor microenvironment, whose heterogeneity affects tumor genesis significantly. Thus, I wanted to assist the current bottleneck on NSCLC treatments by exploring the NSCLC tumor microenvironment and use bioinformatics techniques to discover novel biomarkers to provide new directions to NSCLC drug design and innovation. Ultimately, this led me to hypothesize that there exists undiscovered or untapped biomarkers within the NSCLC tumor micro-environment.
2. What were the major tasks you had to perform in order to complete your project?
The major tasks I had to perform includes (i) standard single cell sequencing analysis for stromal cells, (ii)standard single cell sequencing analysis for epithelial cell-subtypes, (iii) standard single cell sequencing analysis for T-cell subtypes, (iv)InferCNV, (v) functional enrichment analysis, (vi) PPI and Hub Gene calculation, (vi) clinical prognostics analysis, (vii) correlation analysis between immunological components and cancer risk factors, and (viii) protein binding pockets detection.
a. For teams, describe what each member worked on.
I worked alone for this project.
3. What is new or novel about your project?
Both my computational methods and contributions represent novelty in present day NSCLC research. I developed the framework of my project based on the hundreds of literature reviews and journal articles that I referenced. Currently, EGFR, ALK, ROS1, BRAF V600E, MET, RET and NTRK are the major previously identified genetic biomarkers of NSCLC. This study identifies 13 more potential biomarkers.
a. Is there some aspect of your project's objective, or how you achieved it that you haven't done before?
Although I had previous experience with Rstudio coming into the project, conducting this research familiarized me with the tool even more, since approximately 2000 lines of my experiment procedure occurred on Rstudio. Furthermore, while I had a basic understanding of the mechanism of single cell transcriptomics, I have never previously completed a full standard single cell transcriptomics analysis before. To this end, I read over countless literature reviews and single cell RNA sequencing references to familiarize myself with the annotating techniques. Most importantly, I have never read and annotated such a list of extensive and technical papers, analyzed their unique benefits and disadvantages, and planned out a bioinformatics project as I did with this research before. Ultimately, these tasks that I accomplished had significant educational value for me.
b. Is your project's objective, or the way you implemented it, different from anything you have seen?
My project targets NSCLC through the TME (tumor microenvironment) lens, an area with scarce readily available resources. While past researchers have effectively adopted single cell RNA sequencing analysis and used it to identify biomarkers, I provided a unique framework to perform the analysis that I did. Namely, my project thinking combines multiple approaches of analysis, from utilizing various modellings of the TCGA dataset to distinct enrichment analysis methodologies and combining these results with protein binding visualizers like Pymol.
c. If you believe your work to be unique in some way, what research have you done to confirm that it is?
I believe that my results are extremely unique. I identified 13 untapped NSCLC biomarkers and two of which with significant contributions to potential drug discovery. First, I used STRING to find the PPI and employed Cytohubba to select 6 Hub Genes. With the 6 Hub Genes, I conducted a Kaplan-Meier Curve analysis, finding that NDUFB8 is statistically significant to NSCLC survival rate. Next, I used the TCGA clinical dataset to conduct cox proportional hazard regression analysis, LASSO regression analysis, and display the results of the Random Forest Algorithm, and found 12 cancer risk factors. Combined, I discovered a total of 13 NSCLC biomarkers. Furthermore, I performed correlation analysis between the immunological tumor environment and the 13 biomarkers, finding a high correlation between RNASET2 and CD4+ and T-regulatory T cell subtypes.
4. What was the most challenging part of completing your project?
I think that the most challenging part of completing my project was the hand annotating portion, a segment of the standard single cell RNA sequencing analysis. This was because I had limited experiences with excel previous to conducting this project, which meant that I had to filter out marker genes manually. Furthermore, given the huge variety of references, many marker genes can be matched up with multiple cell-subtypes, which makes the process difficult. For example, due to the similarities between T-regulatory cell-subtype and CD4+ cell-subtype, they share similar genetic compositions. Fortunately, the violin graph and heat map for the focus genes that I used validated the significance of expression differences of the marker genes that I hand-picked.
a. What problems did you encounter, and how did you overcome them?
One problem that occurred constantly throughout my project was the size of my dataset. For instance, when I was conducting Copy Number Variation inferences, the dataset that I was using could not be processed through Rstudio, an application that I am mostly proficient and familiar with. This led me to perform certain portions of graphing on Linux instead of Rstudio. While I had to learn Linux from the basics, I overcame my obstacle with data processing by utilizing various online resources from university open-courses as well as Youtube. Moreover, when compiling my codes in Rstudio, the system reported a large number of small errors, and I overcome this obstacle by fixing my code line by line patiently.
b. What did you learn from overcoming these problems?
Technically, this process advanced my skills in both Rstudio and Linux significantly, especially, it built me a solid foundation for Linux, a platform that I have never previously utilized. Similarly, the hand annotating process accustomed me with excel, which contributed significantly to my research also in other sections. Moreover, facing these problems taught me patience, teaching me that there is always a way to defeat bottlenecks. Through trying to figure out these technical difficulties myself, a time consuming but fulfilling process, I became a better researcher and student.
5. If you were going to do this project again, are there any things you would you do differently the next time?
There are three main areas where I would elevate in my project. First, instead of using 19 samples of NSCLC to find statistically significant biomarkers, I would choose an even larger sample size, a process that my new-gained skills in Linux would allow for. A larger sample size will ultimately provide a more accurate and valid overview of the landscape. Second, I would diversify the types of my dataset. In my research, I focused solely on using single cell RNA sequencing. If I were to do this project again, however, I would also utilize bulk analysis, a different type of single cell transcriptomics, to validate my hypothesis. Finally, the main limitation of my study derives from the fact that the 19 samples I selected focus on LUSC, a subtype of NSCLC and not LUAD. This creates a limitation
6. Did working on this project give you any ideas for other projects?
Working on this project opens up a variety of opportunities in NSCLC research. For example, from the two novel immunological biomarkers with protein binding pockets that my research discovered, I can conduct research to create new, distinct algorithms to calculate the ligand compatibility between the protein binding pockets I detected and potential medical treatments. To specify, I can test if potential drug designs (medical inhibitors to limit the expression of RNASET2 and LAMB1 genes) would successfully aid NSCLC clinical treatments. Moreover, I can produce new algorithms to predict the probability of cryptic protein pockets, similar to the PocketMiner graph neural network, and further expand the predicting algorithm from the probability of potential cryptic pockets to their potential shapes (Meller et al., 2023). This experimentation may potentially help me identify whether other NSCLC risk factor biomarkers have protein pockets unidentifiable by PyMol, the data visualization system that I utilized for my research.
7. How did COVID-19 affect the completion of your project?
My project does not require a wet lab design and thus was not affected by COVID-19.